1. Introduction
CrySPY is a software package that searches and optimizes crystal structures based on a given composition. It uses first-principles or molecular dynamics software to evaluate structural energies. In this tutorial, we introduce a calculation using the Docker version of MateriApps LIVE! with Random Search to find the optimal structure for a composition of 12 hydrogen atoms and 12 carbon atoms (expected to yield the benzene structure). The energy evaluation of structures is performed using QUANTUM ESPRESSO, which is pre-installed in MateriApps LIVE!. This article is based on the following tutorial:
https://tomoki-yamashita.github.io/CrySPY_doc/index.html
2. Installation
Run the following command to install CrySPY:
pip3 install csp-cryspy
Many scripts will be installed automatically during this process. Since we will use the cryspy command, check its installation with the following command:
which cryspy
If it is not found, refer to the installation log and add the path to the scripts directory to your PATH environment variable:
echo 'export PATH=$PATH:/home/user/.local/bin' >> ~/.bashrc
source ~/.bashrc
3. Preparing Input Files
Download the benzene example from the official CrySPY manual and place it in a working directory. The directory contents should look like this:
cd qe_benzene_2_RS_mol
tree
.
|-- README
|-- calc_in
| |-- job_cryspy
| |-- pwscf.in_1
| `-- pwscf.in_2
`-- cryspy.in
1 directory, 5 files
Modify the job command in cryspy.in to suit your environment. Here, we use jobcmd = bash.
In job_cryspy, replace /path/to/pw.x with the correct path for your environment. If the path is already set, simply using pw.x is sufficient.
In pwscf.in_1 and pwscf.in_2, specify the directory containing pseudopotentials in the pseudo_dir parameter. In this example, we create a directory named pseudo in the home directory and place the files there: pseudo_dir = '/home/user/pseudo/'.
Next, prepare the pseudopotentials. The files pwscf.in_1 and pwscf.in_2 reference H.pbe-kjpaw_psl.1.0.0.UPF and C.pbe-n-kjpaw_psl.1.0.0.UPF. Download the same files from the official QUANTUM ESPRESSO pseudopotential database and place them in the pseudo directory.
4. Execution
First, specify the number of parallel processes to use:
export NSLOTS=1
While in the qe_benzene_2_RS_mol directory, execute:
cryspy
This will generate six structures. Run cryspy again to evaluate the energies of two structures at a time. Continue executing cryspy until the message Done all structures! appears.
Alternatively, you can use the repeat-execution script for cryspy, which automates this process. The entire computation took approximately six hours and twenty minutes to complete.
5. Results
Check the results in the cryspy_rslt_energy_asc file inside the data directory:
Spg_num Spg_sym Spg_num_opt Spg_sym_opt E_eV_atom Magmom Opt
0 58 Pnnm 58 Pnnm -133.439385 NaN done
2 71 Immm 71 Immm -133.436956 NaN done
3 136 P4_2/mnm 136 P4_2/mnm -133.436394 NaN done
5 53 Pmna 53 Pmna -133.433717 NaN done
4 136 P4_2/mnm 65 Cmmm -133.429064 NaN done
1 132 P4_2/mcm 132 P4_2/mcm -133.427469 NaN done
The structures are listed in ascending order of total energy. Visualizing opt_CIFS.cif with VESTA reveals that the structure with ID 0 (the lowest energy) corresponds to the expected benzene structure, as shown below.
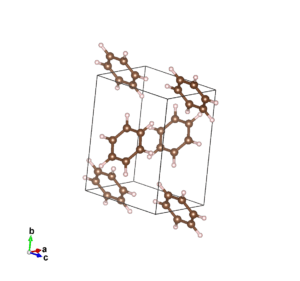
6. Conclusion
In this example, we used Random Search, but CrySPY also supports other structure-search methods such as evolutionary algorithms, Bayesian optimization, and Look Ahead based on Quadratic Approximation (LAQA), which can perform more efficient explorations.